Fabry disease is a progressive genetic condition that causes multiple health problems. Fabry disease occurs when a person’s body does not make enough of an enzyme called alpha-galactosidase A (alpha-Gal) due to changes or mutations in the GLA gene. When alpha-gal is not working, substances called glycolipids build up in the body’s lysosomes (the “recycling centers” of the cell). This storage leads to narrowed blood vessels, inflammation, and health problems all over the body, particularly in the skin, kidneys, heart, brain, intestines and nerves.
Fabry disease is commonly divided into two types, “classic” and “non-classic” or “later onset.” Both men and women can have classic and non-classic Fabry disease. The type of Fabry disease often determines the age symptoms start, the organs affected by the disease, how fast the disease progresses, and how severe symptoms become. Classic Fabry disease symptoms in males and females typically start in the first 2-10 years of life with the onset of burning pain in the hands and feet, decreased sweating, problems in the heat, a reddish-purplish rash, and gastrointestinal issues such as diarrhea, bloating, pain and constipation. Without treatment, the classic form of the disease progresses into kidney disease, heart disease, and increased stroke risk between the ages of 20 to 45. In non-classic Fabry disease, symptoms may start somewhat later in life and may more severely affect one organ like the heart or kidneys. In non-classic Fabry disease, heart disease and other symptoms still occur earlier than average in men than women so it is important to monitor the heart, kidneys, and brain from the time of diagnosis with Fabry disease. In both classic and non-classic Fabry disease, symptoms always worsen over time.
Inheritance of Fabry Disease
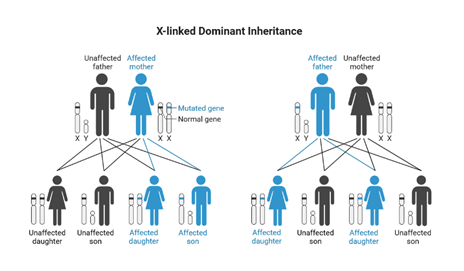
Fabry disease is passed through families in an X-linked inheritance pattern, meaning the GLA gene that causes Fabry disease is located on the X-chromosome. Women have two copies of the X-chromosome (XX) and men have one copy of the X-chromosome and one copy of a Y-chromosome (XY). If men inherit an X chromosome containing the non-working GLA gene, they are unable to produce working alpha-Gal enzyme. Without alpha-Gal, GL-3 builds up in the body and causes the symptoms and health problems of Fabry disease. On the other hand, if women inherit an X chromosome with a nonworking GLA gene, they can have a second X-chromosome with a working GLA gene that can produce alpha-Gal enzyme in some cells of their body. How many cells are able to produce alpha-Gal enzyme in a specific woman depends on X-inactivation, the process by which one of the copies of the X chromosome is turned off in a specific cell before birth. The more X-chromosomes with working GLA gene are turned on, the more enzyme in those cells.
In Fabry disease one copy of the mutation/change in the GLA gene is enough to cause the condition. Females with a mutation on one X chromosome are affected and males with a mutation on their only X chromosome are affected. The children of an affected female, whether male or female, have a 50% chance of being affected. When a male is affected with an X linked dominant condition all of his daughters will have the condition but he will not pass along the gene change to his sons. There is no male to male transmission of an X linked dominant condition.
In the past, it was believed that women who were “carriers” would not have Fabry-related health problems because they had a normal second copy of the gene. However, it is now known that women are not just carriers, and they can and do have Fabry-related health problems. In some cases, women can have health problems as severe as their male relatives. Since women who carry one copy of the non-working gene can have symptoms of Fabry disease, it is important that they discuss Fabry disease with their doctor and obtain appropriate referrals to monitor their health.
Testing for Fabry Disease
The best way to start testing for Fabry is by testing the level alpha-galactosidase A (alpha-gal) enzyme in the blood. If low or missing alpha-gal levels are found, the next step is to identify a disease-causing mutation or change in the GLA gene by gene sequencing analysis to confirm a diagnosis of Fabry disease. Many women with Fabry disease can have normal levels of alpha-Galactosidase A enzyme so it is important for women to have the GLA gene sequencing to establish a diagnosis.
If an individual with health problems suggestive of Fabry disease and/or has low alpha gal enzyme levels with normal GLA gene sequencing results, then there is an additional test called deletion/duplication testing that can pick up missing or added information in the GLA gene that can be missed in normal sequencing genetic testing.If there is a known GLA gene mutation in the family, a person at risk for Fabry disease in that family can have testing for that specific GLA gene mutation.
Treatment for Fabry Disease
In the United States, the only available FDA approved ERT is Fabrazyme (also called agalsidase beta). Outside of the United States, many countries have approved the use of Fabrazyme® and Replagal (agalsidase alfa) for treating Fabry disease. Enzyme Replacement Therapy (ERT) is a specific medication made to replace the normal enzyme that is missing in a person with genetic conditions.
Migalastat or Galafold® is a chaperone therapy pill taken every other day to treat Fabry disease. Migalastat is designed to work on the body’s natural alpha-galactosidase A enzyme by helping make the misfolded enzyme more stable and chaperoning it to the lysosome to do its job. The enzyme should then be able to break down stored glycolipids and prevent them from being stored in cells within the body. Based on studies that look at the gene changes or disease-causing mutations in people with Fabry disease,about 60% of individuals with Fabry disease have amenable mutations which means they are responsive to treatment with migalastat and the drug may be an option for them to treat Fabry disease.
Early intervention with enzyme replacement therapy (ERT) or chaperone therapy offers the best protection against the complications and health problems related to Fabry disease. In adults, this means beginning ERT or oral therapy as soon as Fabry disease is diagnosed. In children with Fabry disease, the decision to begin therapy is based on the symptoms that they have and discussions of the risks and benefits of ERT with your Fabry disease specialist.
Fabry Disease Research
Treatments are being studied to address the underlying causes of Fabry disease that may someday lead to a cure or at least put the disease “into remission”. These treatments include gene therapy, which could add or replace the non-working GLA gene in Fabry patients and help the body make its own alpha-galactosidase A enzyme. Other research includes a gene editing technology called CRISPR that would act as a “spellcheck” and correct the changes or mutation in the non-working GLA gene to a working one. For the most up-to-date list of ongoing clinical trials, please visit clinicaltrials.gov
Fabry Resources
- Fabry Support and Information Group (FSIG)
- National Fabry Disease Foundation (NFDF)
- Fabry Disease- GeneReviews
- Discover Fabry resources and support
- Emory University School of Medicine-Fabry Resources
- NCBI Genetic Testing Register
There are also online communities for rare disorders, such as
Last Updated: 04/21/2022 by Amy Rickheim
Frequently Asked Questions About Fabry disease
Will there ever be a cure for Fabry disease?
A cure for Fabry disease would be a one-time treatment that would prevent all medical issues related to Fabry disease. Currently, we have life-long treatments for Fabry disease, but not a cure.
Treatments are being studied to address the underlying causes of Fabry disease that may someday lead to a cure or at least put the disease "into remission". These treatments include gene therapy, which could add or replace the non-working GLA gene in Fabry patients and help the body make its own alpha-galactosidase A enzyme. Other research includes a gene editing technology called CRISPR that would act as a "spellcheck" and correct the changes or mutation in the non-working GLA gene to a working one. To find additional information about current Fabry disease research or clinical trials, please refer to the Clinical Trials website, talk to the national Fabry support groups, or reach out to your local Fabry treatment center.
Will my insurance cover genetic testing for Fabry disease?
Most health insurance companies will cover most testing costs if you have symptoms or a family history of the condition. Your doctor or genetic counselor might need to write a letter to explain why testing is needed (medically necessary). If you are being tested for Fabry disease through the AAKP/Emory Fabry Testing Project, your testing will be free of charge and your insurance will not be billed for the test. To access the Emory Fabry Testing Project follow the link. Other free or low cost testing options can be found at the discover Fabry website.
Will my daughter with Fabry disease have different symptoms than my son with Fabry disease?
In most cases of classic Fabry disease, boys and girls will present with the same symptoms in childhood. However, because of the way that Fabry disease is passed through families, the severity of symptoms and long term health problems may vary more in girls than in boys. For this reason, it is important that girls have full medical evaluations annually to monitor for symptoms.
In classic Fabry disease, the early signs for both boys and girls usually begin in childhood and can include overheating or sensitivity to hot weather, tingling or pain in the hands and feet, a reddish purple skin rash, protein in the urine, and stomach issues, such as frequent bloating or diarrhea. The most common early symptoms of Fabry disease in kids are a tingling pain in the hands and feet when overheated or sick, and frequent bloating and diarrhea.
Children with non-classic or late onset Fabry disease may also present with some or all of these symptoms and their health should be monitored in either type. Based on the United States expert treatment recommendations, patients of either gender that present with any symptoms due to Fabry disease should be considered for Fabry treatment such as enzyme replacement therapy. Treatment guidelines in other countries may provide insight on when to initiate and cease therapy. In order to find a Fabry disease specialist or more information on Fabry disease treatment, please refer to the National Fabry Disease Foundation online resource.
Will it hurt my baby if I become pregnant during treatment for Fabry disease?
Before becoming pregnant, any woman living with Fabry disease wishing to consider pregnancy should discuss their medications with their doctor and a genetic counselor. Although there are case reports of healthy babies born to mothers on enzyme replacement therapy (ERT), no formal human studies have been done to establish the safety and efficacy of ERT or chaperone therapy for Fabry disease during pregnancy. Accordingly, Fabry specialists and obstetrician/gynecologists (ob/gyn) will work closely together to determine when to stop and start Fabry specific medications during pregnancy.
Other drugs that a woman may be taking to manage symptoms of Fabry, such as Dilantin (Phenytoin) or Tegretol (carbamazepine) for hand and foot pain, may also increase the risk of certain birth defects and developmental delays in an unborn baby. If a woman with Fabry disease becomes pregnant unexpectedly, it is important to consult with a doctor as soon as possible to discuss medication use options.
For more information about Fabry disease and pregnancy, please talk to your Fabry doctor or genetic counselor. A Fabry focused healthcare provider can be found on the National Fabry Disease online resource. To find a genetic counselor in your area, use the National Society of Genetic Counselors Find a Genetic Counselor tool on the National Society of Genetic Counselors website.
Will Fabry affect cognitive function, even without strokes?
People living with Fabry disease do not have intellectual impairments or learning problems more frequently than average people without Fabry disease. However, they are at higher risk for mini-strokes, TIAs (transient ischemic attacks), and strokes due to the disease’s effect on the brain. In addition to damage from strokes, some adults with Fabry disease do report symptoms such as "fuzzy thinking," occasional decreased memory for names and dates, and attention deficit issues, even in the absence of stroke damage. There is evidence that some individuals with Fabry disease have issues with executive functioning (verbal generation, reasoning, problem solving, perseveration), information processing speed, and attention from testing, but the exact cause is unknown. Research on brain functioning in Fabry disease has not discovered an accurate method to measure "fuzzy thinking," despite trying through a variety of neurocognitive testing studies. Dr. Nadia Ali, a health psychologist at the Emory University Lysosomal Storage Disease Center in Atlanta, GA is very interested in learning more about this issue and is conducting research on "fuzzy thinking" in Fabry disease. She can be contacted directly for more details: Dr. Nadia Ali’s faculty page.
Why is there fatigue with Fabry disease?
The exact causes for the chronic fatigue and exhaustion in Fabry disease are unknown. It may be related to the disease burden on the body’s function, depression, poor sleeping, and/or heart rhythm issues. We do know that many individuals with Fabry disease do report having more energy after several months of enzyme replacement therapy every two weeks. Other strategies to manage fatigue include lifestyle changes to conserve energy and include taking frequent naps, avoiding certain activities, being prepared for changing weather conditions, and increasing water or liquid consumption. If a person diagnosed with Fabry disease is experiencing extreme fatigue, it is important for them to talk to their doctor about secondary causes such as low thyroid function, low Vitamin B levels, low Vitamin D levels, sleep apnea, or heart disease. Some individuals may also be interested in talking to their doctor about taking medications such as Provigil (modafinil) that can address some chronic fatigue issues.
Why does it take so long for some people to be diagnosed with Fabry disease?
Fabry disease can take a long time to be diagnosed because the symptoms are not very specific and they overlap with other more common diseases, like chronic fatigue syndrome and irritable bowel syndrome. People may be called hypochondriacs or told symptoms are "all in their head" because many standard lab tests and assessment can’t detect Fabry disease. Fabry disease is considered a rare disease and it is often not in the list of possible causes for health problems. Many people with Fabry disease are given a symptom specific diagnosis or a misdiagnosis until an eye finding, a kidney biopsy, or a family member is diagnosed with Fabry disease leading to the right diagnosis.
Why does Fabry disease cause pain in the hands and feet?
In Fabry disease, the build up of GL-3 and related lipids damages small nerve fibers and interferes with normal functioning of nerve cells. Individuals often experience itching, numbness, tingling, burning, pain, sensitivity to cold, and problems sweating in their hands and feet due to the damage. Medical professionals and doctors may also refer to this as acroparesthesias or neuropathy. Enzyme replacement therapy (ERT) and/or prescription medications such as diphenylhydantoin, carbamazepine and gabapentin have been successful in some individuals living with Fabry disease in alleviating nerve pain.
There are many ways that individuals living with Fabry disease may describe the pain they feel in their hands and feet. Itchy feet, sand in the shoes, dragon fire in the feet and hands, frostbite, and other terms have all been used to describe this type of pain.
Why do I have to take Galafold every other day?
Galafold works by helping to stabilize the faulty enzyme in Fabry disease and guiding it into the lysosomes to help break down accumulated lipid waste. Therefore, it is called a chaperone therapy. Once the enzyme has entered the lysosome, the chaperone will detach from the enzyme so it can break down the waste. The every-other-day dosing allows time for the protein to detach and let the enzyme complete its work. This is a case where more is not necessarily better.
Who is Fabry disease named after?
Fabry disease is named after the first doctor, Dr. Johannes Fabry, who described the signs and symptoms of the disease in 1898. Fabry disease is also sometimes referred to as Anderson-Fabry disease, acknowledging another doctor, Dr. William Anderson, who also described its symptoms in 1898. Both Dr. Fabry and Dr. Anderson were dermatologists, or specialized doctors that treat skin problems. They both had patients with skin lesions common to Fabry disease called angiokeratomas. "Angio" refers to blood vessels, and "keratoma" refers to hardened or callous skin. Angiokeratomas look like a painless, red rash and usually appear around the "bathing truck" area of the body or around the elbows and knees.
Where is the Fabry Center in Georgia?
The Emory Fabry and Lysosomal Storage Disease Center (Emory LSDC)and Emory Genetic Clinic Trials Center (Emory GCTC) in Atlanta, Georgia provide diagnostic, evaluation, management, research, and treatment services for Fabry patients from all over the Southeastern United States. The Emory LSDC is devoted to remaining on the cutting edge of research and treatment providing comprehensive and compassionate care for all of our patients affected by Fabry disease. The Emory GCTC is dedicated to compassionately working with Fabry patients to provide access to innovative clinical studies in Fabry disease across the United States. To schedule an appointment or speak with a member of Emory Fabry team, call 404-778-8518 or 800-200-1524 or email Dawn Laney, MS at dawn.laney@emory.edu. You can also visit the Emory LSDC website or Emory GCTC for more information.
Where do I learn more about clinical trials for Fabry disease?
The easiest way to learn more about studies and clinical trials in Fabry disease is to talk to a genetic counselor, a doctor that specializes in Fabry disease, or medical geneticist that is familiar with Fabry disease. However, studies focusing on treatments and cures are also posted at clinicaltrials.gov where you can search for "Fabry disease." It may help to avoid limiting your search to your state or country, as many studies will pay for your travel if you are eligible to join.
Another great resource for information on studies are the Fabry support groups. The support websites that may be of help are: The National Fabry Disease Foundation, Fabry Support and Information Group or FSIG
Individuals interesting in learning more about what it means to participate in a clinical trial may wish to visit webpages such as the Emory Genetic Clinical Trials Center Clinical Trials Resources Page to learn more about being a part of these studies.
Antidote also offers a handy tool to find clinical trials.
Where can I find out more information on Fabry disease and enzyme replacement therapy?
The best source of information on enzyme replacement therapy (ERT) is a doctor specializing in Fabry disease. The National Fabry Disease Foundation has a Fabry specialist finder on their website. Additional information can be obtained from either a Fabry support group or informational websites sponsored by the companies that make ERT. The international organization FIN may also be great resource for those living outside the United States.
Where can I find a treatment center for Fabry disease?
Fabry disease and Lysosomal Storage Disease Centers (LSDCs) are genetic centers that specialize in the treatment of patients with Fabry disease. Most centers have a medical geneticist, genetic counselor, kidney doctor (nephrologist), heart doctor (cardiologist), and nurse who work as a team to answer your questions, discuss testing, identify your at-risk family members, and develop a comprehensive evaluation and treatment plan for you. The Fabry specialist will work with your current doctors to organize the treatment, tests, and specialists you need. There is a Fabry clinic finder at the National Fabry Disease Foundation website.
The Fabry International Network is a patient advocacy group that can help identify Fabry specialist centers to patients living outside the United States.
Where can I call to learn more about Fabry disease?
A Fabry center near you can be a great resource. The National Fabry Disease Foundation has a Fabry specialist finder on their website. Other websites that may be of help are:
When should we begin enzyme replacement therapy for our children with Fabry Disease?
The timing of initiation for enzyme replacement therapy (ERT) can vary depending on the wishes of the family, the child’s symptoms, and the opinions of the doctor. Often, the treating physician will closely monitor gastrointestinal, kidney, and heart functions while also asking questions about the child’s energy and pain. They often recommend beginning ERT when symptoms of the disease emerge and begin to affect daily life and when symptoms begin to progress. The parents’ wishes are always taken into account when determining the time to begin therapy. At this time, therapy rarely is started before three years of age. Please talk to your Fabry doctor or genetic counselor to come up with the best treatment and monitoring plan.
When should I test my children to see if they have Fabry disease?
Some families choose to test their children before birth through prenatal testing. Some families test for Fabry disease at birth or later when their children can understand the implications of the test. No matter when you choose to test, there are special issues to consider prior to testing such as:
When should ERT be started for Fabry disease?
Experts agree ERT should be started as early as possible. Early intervention with enzyme replacement therapy (ERT) offers the best protection against the complications and health problems related to Fabry disease. In adults, this means beginning ERT as soon as Fabry disease is diagnosed. In children with Fabry disease, the decision to begin therapy is based on the symptoms that they have and discussions of the risks and benefits of ERT with their Fabry disease specialist.
In the United States, many experts recommend treating males and females with classic and non-classic Fabry disease as soon as they have symptoms, such as tingling or pain in their hand or feet or issues with diarrhea and constipation. In people without symptoms, the doctor will look at the family history of Fabry symptoms and the gene change causing Fabry disease and work with them to make a good plan for watching for Fabry disease symptoms and starting treatment. For more information about Fabry disease treatment and ERT, please talk to your Fabry doctor or refer to the National Fabry Disease online resource.
When can the Fabry infusion nurses increase the speed of my infusion?
If you reach the 7th infusion with Fabrazyme and haven’t had any problems, your doctor and infusion nurse may be able to begin slowly increasing the infusion rate. If you have an infusion related reaction, the time of infusion will be decreased and the speed of the infusion decreased again. After many months of therapy and a gradual increase in the infusion rate, many individuals with Fabry disease can have their infusion over 2 hours.
The manufacturer for Replagal (Shire) does not recommend infusing this product any quicker than 40 minutes.
Please discuss any questions about ERT treatment with your Fabry doctor.
What tests are used to monitor Fabry disease?
Although different Fabry specialists and centers may vary slightly in their test and time schedule. Current accepted and recommended global studies to evaluate Fabry-related symptoms exist and may be performed on an annual basis:
What tests are recommended for monitoring children with Fabry disease?
Children who are diagnosed with Fabry disease should have a detailed physical exam and annual laboratory tests to measure their overall health. Your Fabry doctor will talk to you about important tests and timing for your child. Tests may include:
What tests are performed to diagnose Fabry disease?
Fabry disease can be diagnosed through a simple blood or saliva test. In males, the best way to start testing is doing alpha-galactosidase A (alpha-gal) enzyme testing on blood. If low or missing alpha-gal levels are found, the next step is to identify a disease causing mutation or change in the GLA gene by gene sequencing analysis to confirm a diagnosis of Fabry disease. Many women with Fabry disease can have normal levels of alpha-Galactosidase A enzyme so it is important for women to have the GLA gene sequencing to establish a diagnosis.
If an individual with health problems suggestive of Fabry disease and/or has low alpha gal enzyme levels with normal GLA gene sequencing results, then there is an additional test called deletion/duplication testing that can pick up missing or added information in the GLA gene that can be missed in normal sequencing genetic testing.
If there is a known GLA gene mutation in the family, a person at risk for Fabry disease in that family can have testing for that specific GLA gene mutation. A list of laboratories that perform testing for the GLA gene can be found in the NCBI Genetic Testing Registry. If testing is being delayed by lack of insurance or large out of pocket payments, there are free and reduced cost testing options in Fabry disease. Contact the Fabry support groups, Fabry Support and Information Group (FSIG) and the National Fabry Disease Foundation (NFDF), or a center specializing in Fabry disease care to learn more about these options. In order to find a Fabry disease specialist or more information on Fabry disease treatment, please refer to the National Fabry Disease Foundation online resource.
Resources for Fabry disease outside the United States may be available by speaking to your clinician or patient advocacy group.
What symptoms might indicate my kidney function is worsening?
People in the early stages of kidney disease usually do not feel sick at all. The first sign of a kidney problem may be high blood pressure, a low number of red blood cells (anemia), or blood or protein in the urine (proteinuria). If kidney disease gets worse, people may need to urinate more or less often. They may feel tired or itchy. They may lose their appetite or experience nausea and vomiting. Their hands or feet may swell or feel numb. They may get drowsy or have trouble concentrating. The person’s skin may darken. They may have muscle cramps. Please contact a Fabry doctor and/or kidney doctor immediately if an individual appears to be experiencing any of these symptoms. In order to find a Fabry disease specialist or more information on Fabry disease and the kidneys, please refer to the National Fabry Disease Foundation online resource.
What states offer newborn screening for Fabry disease?
In the United States, there is newborn screening for Fabry disease in several states. These states do testing for enzyme levels of alpha-galactosidase A, or alpha-Gal, on newborn dried blood spots. Currently Missouri, Illinois, Oregon, Tennessee, and Maryland screen all babies born in the state for Fabry disease. Pennsylvania and New York offer the option for Fabry disease testing on the newborn screening panel(in selected areas of New York City). New Jersey and New Mexico will soon offer newborn screening for Fabry disease. Not all states test for Fabry disease as it has not yet been added to the Recommended Uniform Screening Panel (RUSP) that many states use to decide which new tests to begin adding to their newborn testing. A current listing of states performing newborn screening for Fabry disease can be found on the NewSTEPs website in the data visualizations section.
It is important to remember that a "positive" newborn screen for Fabry disease does not mean that a child has the condition. A false positive can occur for many different reasons and there are important follow up tests that need to be done to determine if a child actually has Fabry disease.
For more information about newborn screening for Fabry Disease, please refer to the Baby’s First Test online resource.
In order to find a Fabry disease specialist who can provide more information on newborn screening for Fabry disease, please refer to the National Fabry Disease Foundation online resource.
What should I tell the school about my child’s Fabry disease?
Teachers, particularly physical education teachers and coaches, should understand the symptoms of Fabry disease and how a child is functioning physically. A child’s teachers should be informed about any medications and the special needs of Fabry patients, such as the necessity to avoid over-heating during times like recess or physical education. Make sure that the school has updated medical files and explain expectations if a child gets ill at school. It is important to emphasize that the children with Fabry disease do not have an increased risk for learning disabilities and that Fabry disease cannot "infect" other children.
For more information about how to talk to your child’s teacher or school about Fabry disease, please refer to the Fabry Support and Information Group or the National Disease Foundation. For further resources and books that can help explain Fabry disease to classmates and teachers, please visit the Emory Fabry Center’s resources webpage.
What should I tell my pediatrician about Fabry disease?
Pediatricians and Fabry doctors should work together as a team to treat children living with Fabry disease. When working with a new pediatrician, parents should take the time to explain the disease as much as possible and encourage him or her to speak to a genetic specialist with any questions. They should understand the possible complications of Fabry disease, so they can monitor for complications during their regular examinations or any sick visits. There are several useful medical articles that Fabry specialists can provide pediatricians about the symptoms and treatment of Fabry disease in children such as this guideline to taking care of children with Fabry
Fabry specialists can provide pediatricians with information about the symptoms and treatment of Fabry disease in children. The following article is a comprehensive guide for the management and treatment of children with Fabry disease: The management and treatment of children with Fabry disease: A United States-based perspective.
What other kind of specialists will I need to see with Fabry disease?
Patients affected by Fabry disease should visit their primary care physician and Fabry doctor at least once a year for a thorough evaluation, physical exam, and laboratory tests. These tests help monitor disease progression and overall health. Many individuals affected by Fabry disease will also have a nephrologist to monitor kidney functions, a cardiologist to monitor their heart, a neurologist to monitor their brain function and pain, and a psychologist to monitor depression or anxiety. Other specialists may be needed to address gastrointestinal issues or other Fabry related symptoms. Please talk to your Fabry doctor or genetic counselor for more information about other specialists that you might need to see.
What is the usual abbreviation for Fabry disease?
The usual abbreviations for Fabry disease are FD or AFD.
What is the life expectancy of someone with Fabry disease?
Fabry disease (FD) is a progressive condition that causes medical issues throughout the body, including a negative impact on the function of the kidney and heart. Before kidney transplants were used to treat patients with Fabry disease, life expectancy was directly related to how well the kidneys were functioning. Accordingly, individuals may find information suggesting that men with classic Fabry disease have a very reduced life expectancy. When kidney transplants became available, the time to death improved from an average life expectancy to around 58.2 years for men and 75.4 years for women as reported in 2009. However, the availability of enzyme replacement therapy (ERT), access to earlier kidney transplants, improvements in understanding of disease complications, and the increased awareness of heart rhythm abnormalities and the need for aggressive treatment have changed life expectancy in Fabry disease for the better. Preliminary evidence suggests that earlier treatment with ERT greatly reduces the impact of Fabry disease on the body and quality of life on men and women with classic and non-classic Fabry disease. It will take time to understand how treatment will change life expectancy in Fabry disease, but there are men on ERT with classic Fabry disease living past 60 years of age with good quality of life.
What is the difference between classic and non-classic Fabry?
Fabry disease can be associated with a wide range of severity and it is commonly divided into two types, "classic" and "non-classic or late onset." Both men and women can have classic and non-classic Fabry disease. Because of the way Fabry disease is passed through families, symptoms often vary more in women than in men. The type of Fabry disease often depends on the age symptoms start, the organs affected by the disease, how fast the disease progresses, and how severe symptoms become.
Classic Fabry disease symptoms in males and females typically start in the first 2-10 years of life with the onset of burning pain in the hands and feet, heat intolerance and gastrointestinal issues, such as diarrhea, pain and constipation. Without treatment the classic form of the disease progresses into the kidneys, heart, and causes other health problems to occur between the ages of 20 to 45.
In non-classic Fabry disease, symptoms may start after childhood and may more severely affect one organ like the heart or kidneys. It is important to note that in non-classic Fabry disease, heart disease and other symptoms still occur earlier than average in men than women, so it is important to monitor the heart, kidneys, and brain from the time of diagnosis with Fabry disease. In both classic and non-classic Fabry disease, symptoms always worsen over time.
Rarely, Classic Fabry disease will be referred to as "Type I" and Non-Classic Fabry disease will be referred to as ‘Type II" but that is not the preferred terminology.
To learn more about classic and non-classic Fabry disease, please talk to your doctor or speak with a Fabry center team member. Find a Fabry center near you at the National Fabry Disease online resource.
What is the connection between Fabry disease and pirates?
When newborn screening for Fabry disease began in Taiwan, the testing found an unexpectedly high number of babies with a late-onset Fabry-causing pathogenic change or variant in the GLA gene called c.640-801GtoA (also known as IVS4+919GtoA and c.639+919GtoA). Further research in the ancestry of these families found that the gene change was originally brought to the island of Taiwan by a group of seafaring Han Chinese pirates led by Chief pirate Wang Zhi in the 16th century. It is believed that this group traveled through several ports and countries and expanded the population of individuals with Fabry disease throughout the region.
What is Fabry disease?
Fabry disease is a progressive genetic condition that causes health problems like extreme tiredness (fatigue), little to no sweating (hypohidrosis), whorls in the cornea of the eyes (cornea verticillata), reddish-purplish skin rashes (angiokeratomas), burning pain in the hands and feet (neuropathic pain), problems in the heat and cold, frequent diarrhea and constipation, protein in the urine, and slow heartbeat (bradycardia). Without treatment, Fabry-related health problems will worsen leading to heart disease, kidney disease, and increased risk for strokes. Fabry disease occurs when a person’s body does not make enough of an enzyme called alpha-galactosidase A (alpha-Gal) due to changes or mutations in the GLA gene. When alpha-gal is not working, substances called glycolipids [globotriaosylceramide (GL-3 or GB3) and lyso-globotriaosylceramide (Lyso-GL3 or LysoGB3)] build up in the body’s lysosomes (the "recycling centers" of the cell). This storage leads to narrowed blood vessels, inflammation, and health problems all over the body, particularly in the skin, kidneys, heart, brain, intestines and nerves.
Fabry disease can be associated with a wide range of symptoms but is commonly divided into two types, "classic" and "non-classic" or "later onset." Both men and women can have classic and non-classic Fabry disease. Because of the way Fabry disease is passed through families, symptoms often vary more in women than in men. The type of Fabry disease often depends on the age symptoms start, the organs affected by the disease, how fast the disease progresses, and how severe symptoms become. Classic Fabry disease symptoms in males and females typically start in the first 2-10 years of life with the onset of burning pain in the hands and feet, decreased sweating, problems in the heat, a reddish-purplish rash, and gastrointestinal issues such as diarrhea, bloating, pain, and constipation. Without treatment, the classic form of the disease progresses into kidney disease, heart disease, and increased stroke risk between the ages of 20 to 45. In non-classic Fabry disease, symptoms may start somewhat later in life and may more severely affect one organ like the heart or kidney. In non-classic Fabry disease, heart disease and other symptoms still occur earlier than average in men than women, so it is important to monitor the heart, kidneys, and brain from the time of diagnosis with Fabry disease. In both classic and non-classic Fabry disease, symptoms always worsen over time.
Currently, there is no cure for Fabry disease; however, there are two Food & Drug Administration (FDA) approved medications in the United States that can help stabilize or prevent the progression of the more serious health problems if started early in the disease’s course. Fabrazyme®, or agalsidase beta, is an enzyme replacement therapy (ERT) usually given every two weeks by IV to break down the stored glycolipids in the body. Fabrazyme® is a treatment that can be given to individuals with any mutation or change in the GLA gene. Migalastat or Galafold® is a pill taken every other day that is designed to help an individual with Fabry disease’s own enzyme work more effectively (chaperone therapy). Galafold® only works for specific mutations in the GLA gene that are considered "amenable". Individuals with Fabry disease can talk to their doctors to learn if they may be able to take Galafold and check to see if their GLA change is considered amenable at the GLA mutation search website.
Outside of the United States, a second enzyme replacement therapy (ERT) medication for Fabry disease is available called Replagal® (agalsidase alfa). Replagal® is a treatment that can be given to individuals with any mutation or change in the GLA gene.
There are symptom specific treatments available to help relieve some of the other health problems of Fabry disease and there are also support networks to help individuals cope with the day-to-day struggles of living with Fabry disease such as the Fabry Support and Information Group (FSIG) and the National Fabry Disease Foundation (NFDF).
There is an excellent summary about Fabry disease written for doctors at Fabry Disease- GeneReviews.
What is enzyme replacement therapy for Fabry disease?
Enzyme Replacement Therapy (ERT) is a specific medication made to replace the normal enzyme that is missing in a person with genetic conditions. Fabry disease is caused by a deficiency or misfolding of the enzyme alpha-Galactosidase A, or alpha-Gal. Alpha-Gal is one of several enzymes in the body’s lysosomes or "recycling centers" responsible for breaking down large substances called glycolipids such as globotriaosylceramide, also called GL-3 or Gb3. Most importantly when alpha-Gal is low, missing, or misfolded, GL-3 builds up in lysosomes throughout the body interfering with normal functioning, causing pain, causing inflammation, and narrowing blood vessels.
In Fabry disease, the replacement enzyme is injected intravenously (IV) every other week and taken up by the cells. Once the medication enters the cells, the enzyme can help breakdown the stored GL-3 and help the cells work better. ERT should stop GL-3 from building up and hopefully slow or stabilize Fabry disease symptoms and health-related problems from getting worse. ERT is not a cure for Fabry disease and needs to be given at least every two weeks as the body metabolizes the replacement enzyme quickly.
In the United States, the only available FDA approved ERT is Fabrazyme (also called agalsidase beta). Outside of the United States, many countries have approved the use of Fabrazyme® and Replagal (agalsidase alfa) for treated Fabry disease.
Chaperone therapy, an oral medication that addresses the lack of Alpha-gal seen in Fabry disease through a different mechanism is also approved for the treatment of Fabry disease inside the United States.
What is a Port-A-Cath and should I get one for my Fabry infusions?
A Port-A-Cath is a combination of a port and an intravascular device used to get intravenous or IV access in the same spot every time for infusions like enzyme replacement therapy (ERT). The device is located under the skin in the upper part of the chest or other spot connected into a large vein. Surgery, under general anesthesia, is usually needed to place the Port-A-Cath. The Port-A-Cath is not required for infusions, but the benefit of the Port-A-Cath is that it allows for an easy access to a vein for infusions. The downsides include: Port-A-Caths are placed through surgery, which requires general anesthesia, there is a risk of infection with the Port-A-Cath, and although Port-A-Cath can last for years, they may need to be replaced or taken out. Usually Port-a-Caths are put in by general surgeons or interventional radiologists. The decision to get a Port-A-Cath is a personal choice that should be discussed with your physician. If you have questions or would like to learn more about Port-A-Caths, please talk to your Fabry doctor or genetic counselor.
What if I have any unusual symptoms after the ERT infusion for Fabry disease?
Some people with Fabry disease feel bloated, tired, have increased leg pain, headache, increased tiredness, chest pain, racing heart, chills, nausea, fever, or itching after their infusion. If you are having these or other health issues after your infusion, please let your health care team and infusion nurse know right away. It may be that taking a medication like Benadryl or a steroid may be needed before the next infusion. Your healthcare team will let you know if you will be taking new medications before your next infusion, have blood collected for antibody testing, and if the infusion will be slowed down next time.
What happens if you stop ERT for Fabry disease?
There are not any detailed studies available that can tell us exactly what happens when you stop enzyme replacement therapy (ERT). We do know that as soon as ERT is stopped, the GL-3 levels begin accumulating again. As per patient reports during a period of ERT medication shortage, we know that patients with Fabry disease often start having Fabry problems, such as diarrhea and extreme tiredness, within a month without ERT. Specific information on how long it takes GL-3 to build up again to previous levels is not available in Fabry disease patients. Studies looking at another lysosomal storage disease, Gaucher disease, found that when ERT is stopped, the storage products built up again quickly in most individuals. Those individuals had a quick regression to the same symptoms they had before beginning ERT. When restarting ERT, it took time to return back to the healthy levels they reached on ERT before stopping. For more information about Fabry disease treatment and the importance of staying consistent with ERT, please talk to your Fabry doctor or genetic counselor.
What happens if I have an infusion-related reaction to enzyme replacement therapy for Fabry disease while at home?
The following content is sponsored by Optum Specialty and Infusion Pharmacies
Home infusion of enzyme replacement therapy (ERT) for Fabry disease follows the same protocols as treatment in a physician’s office or infusion clinic – including management of any infusion-associated reactions. If a patient has a reaction during or after their infusion, nurses will follow the infusion-reaction orders that are in place, administer the appropriate medications, call the managing doctor, and monitor vital signs until the reaction resolves. If a reaction worsens or is not resolving, the nurse will usually call 911 and ensure the patient gets emergency treatment.
If patients or families have questions about handling potential infusion reactions at home, they should always feel free to ask the home infusion nurse or doctor. In addition, some infusion pharmacies, such as Optum Specialty and Infusion Pharmacies, have clinical teams who can walk patients and family members through the step-by-step, infusion-associated reaction management process and answer questions along the way.
What happens during an ERT infusion for Fabry disease?
Depending on the ERT product selected, the first infusion takes approximately 4 hours of actual infusion time and 1 hour after the infusion has ended for monitoring. After several months, the time of infusion can typically be reduced down to 2 hours. You may wish to bring books, a laptop computer, cell phones, etc. A guest/family member/friend may accompany you for your infusion. Depending on where you are having the infusion, you may bring your own lunch, or have a guest pick up lunch for you during the infusion. Please note that if you are infused within a cancer infusion center, they may have restrictions on hot or strong smelling foods. If you receive your infusions at an alternative site, please check with them regarding their food/drink policy.
Before leaving your infusions, make sure to schedule your next infusion. Your infusions should be scheduled every other week.
Please talk to your Fabry doctor or genetic counselor for more information about what to expect with ERT treatment and how to prepare for your first infusion.
What gene change causes Fabry disease?
Fabry disease is caused by changes or mutations in the GLA gene. The mutations in the GLA gene prevent the body from making enough of a working enzyme called alpha-galactosidase A, or alpha-Gal. When alpha-Gal is not working, substances called glycolipids [globotriaosylceramide (GL-3 or GB3) and lyso-globotriaosylceramide (Lyso-GL3 or LysoGB3)] build up in the body’s lysosomes (the "recycling centers" of the cell). This storage leads to narrowed blood vessels, inflammation, and health problems all over the body, particularly in the skin, kidneys, heart, brain, intestines and nerves.
Fabry disease is passed through families in an X-linked inheritance pattern, meaning the GLA gene that causes Fabry disease is located on the X-chromosome. Women have two copies of the X-chromosome (XX) and men have one copy of the X-chromosome and one copy of a Y-chromosome (XY). If men inherit an X chromosome containing the non-working GLA gene, they are unable to produce working alpha-Gal enzyme. Without alpha-Gal, GL-3 builds up in the body and causes the symptoms and health problems of Fabry disease. On the other hand, if women inherit an X chromosome with a nonworking GLA gene, they can have a second X-chromosome with a working GLA gene that can produce alpha-Gal enzyme in some cells of their body. How many cells are able to produce alpha-Gal enzyme in a specific woman depends on X-inactivation, the process by which one of the copies of the X chromosome is turned off in a specific cell before birth. The more X-chromosomes with working GLA gene are turned on, the more enzyme in those cells.
In the past, it was believed that women who were "carriers" would not have Fabry-related health problems because they had a normal second copy of the gene. However, it is now known that women are not just carriers, and they can and do have Fabry-related health problems. In some cases, women can have health problems as severe as their male relatives. Since women who carry one copy of the non-working gene can have symptoms of Fabry disease, it is important that they discuss Fabry disease with their doctor and obtain appropriate referrals to monitor their health.
In order to find a Fabry disease specialist or more information on Fabry disease treatment, please refer to the National Fabry Disease Foundation online resource.
What family considerations are there for being tested for Fabry disease?
When going through the testing process, you may discover things you did not know about your family. You may learn that your children are at risk of being affected by Fabry disease. This may cause emotional turmoil for yourself and your family. A parent may feel guilty for having "passed Fabry disease" on to their children, even if they know that they have no control over which gene is inherited. Even though they do not want the health issues associated with Fabry disease, people in the family who are found not to have Fabry may feel guilty about having "escaped" the disease while others in the family weren’t as lucky. This is termed survivor guilt. They may even feel left out or isolated on the outskirts of the family as the affected family members all share treatments, support, and physicians. Please talk to your doctor or genetic counselor to discuss the implications for both yourself and your family before testing. Take your time when deciding to get tested for Fabry disease.
For more information about online brochures about Fabry disease or how to get tested for Fabry disease, please refer to the Discover Fabry online resource.
A genetic counselor can help you think about the issues surrounding testing for Fabry disease. Genetic counselors can be found on the [link url=" https://findageneticcounselor.nsgc.org/” target=”_blank”>National Society of Genetic Counselors website.
What effect does Fabry disease have on my kidneys and their function?
Fabry disease is a genetic condition caused by a deficiency of an enzyme called alpha-galactosidase A and mutations in the GLA gene. Alpha-galactosidase A, or alpha-Gal, is one of several enzymes in the body’s lysosomes or "recycling centers" responsible for breaking down substances in the body called glycolipids such as globotriaosylceramide, also called GL-3 or Gb3. Most importantly, when alpha-Gal is low or missing, GL-3 builds up in lysosomes throughout different body systems, results in inflammation, and interferes with normal functioning.
The kidney problems caused by Fabry disease are due to the build up of GL-3 or Gb3 and other similar products in the cells of the kidney. The build-up of GL-3 causes the kidneys to lose their ability to filter waste and chemicals in your body. Without treatment for Fabry disease, the accumulation of these substances can gradually lead to total and permanent kidney failure called end-stage renal disease (ESRD). People with ESRD must undergo dialysis or transplantation to prolong their lifespan. Individuals with Fabry disease are candidates for kidney transplants and should not be denied that option based on their genetic condition alone.
Individuals with Fabry disease often have protein in their urine (proteinuria). Proteinuria is a sign of kidney disease but can also put stress on the kidneys so it is important to seek the advice of a kidney doctor to decrease the levels of protein in the urine as much as possible using medications such as ACE inhibitors (angiotensin-converting enzyme inhibitors) or ARBs (angiotensin-receptor blockers).
Studies have shown that starting enzyme replacement therapy earlier allows for more effective removal of GL-3 from the kidneys, which aids in stabilizing the disease progression.
What dose of Galafold should I take?
The adult dosage of Galafold (migalastat) is 123 mg orally, every other day. The dose should be consumed at the same time of day on an empty stomach. Food should not be consumed 2 hours before and 2 hours after a meal. Capsules should be swallowed whole; do not chew, crush or cut capsules open.
What do people do to treat the gastrointestinal issues seen in Fabry disease?
The GI symptoms in Fabry are thought to be caused by the storage of globotriaosylceramidee (GL-3) in the body that interferes with nerve and cell function in the GI system. This disrupts how fast the stomach empties, how quickly food moves through the intestine, and other functions of digestion including absorbing nutrients. Treatment with enzyme replacement therapy [Fabrazyme (agalsidase beta) or Replagal (agalsidase alfa)] or chaperone therapy [Galafold (migalastat)] has been shown to help decrease the severity and frequency of GI symptoms.
After an evaluation by a gastroenterologist and the ruling out other non-Fabry related causes of GI issues, doctors may also try symptom-specific medications such as: Reglan (metoclopramide) to help the stomach empty, Zofran (Ondansetron) to reduce nausea and vomiting, pancreatic enzymes to aid digestion, Loperamide (Imodium) to decrease hyperactive contractions in patients with diarrhea, or Amitriptyline to decrease nerve pain and other issues in the GI system. Some individuals with Fabry also find that eating small meals, taking probiotics, and avoiding spicy, lactose-containing, or greasy foods also help decrease GI issues.
Dr. Claire Zar-Kessler at Massachusetts General Hospital is also enrolling in a study to learn more about gastrointestinal issues in Fabry disease.
What do I do if I think I am depressed, anxious, or having panic attacks due to Fabry disease?
Please seek help if you think you are depressed, anxious or having panic attacks. Help can be found from your physicians and your family. Remember that depression and anxiety can make some people feel like giving up. It is important to realize that these negative views are part of the disease and may not accurately reflect the actual circumstances. Studies have shown that treatment for Fabry disease does improve quality of life as well as physical health and combining Fabry specific therapy with psychology care can help improve aspects of life. Some Fabry centers have psychologists skilled in evaluating or treating Fabry related depression, anxiety, and panic attacks. Additional information about support groups and strategies for coping with the day-to-day challenges of living with Fabry disease can be found on the Discover Fabry Support online resource.
What causes the gastrointestinal issues seen in Fabry disease?
The exact reason for gastrointestinal (GI) issues, like cycles of chronic diarrhea and chronic constipation, stomach cramping and pain, bloating, nausea, vomiting, and feeling full early or early satiety, in Fabry disease is still unknown. However, doctors feel that these symptoms are likely due to the inability to move food effectively through the GI system. Doctors and medical professionals may refer to this as gastrointestinal dysmotility (GD). It is possible that the GD in Fabry disease is caused by the storage of globotriaosylceramide (GL-3) and other glycolipids that damages the small nerves that help tell the body to move food through the digestive system. This disrupts how fast the stomach empties, how quickly food moves through the intestine, and other functions of digestion (e.g. absorbing nutrients). Various cell types in patients affected with Fabry disease (e.g. nerve, muscle, blood vessel lining, immune, and epithelial cells) were found to store GL-3, which may explain the GI issues presented. Treatment with enzyme replacement therapy [Fabrazyme (agalsidase beta) or Replagal (agalsidase alfa)] and chaperone therapy [Galafold (migalastat)] have been shown to help decrease the severity and frequency of GI symptoms. After a gastroenterologist has ruled out other non-Fabry related causes of GI issues, doctors may also try symptom specific medications such as: Reglan (metoclopramide) to help the stomach empty, Zofran (Ondansetron) to reduce nausea and vomiting, pancreatic enzymes to aid digestion, Loperamide (Imodium) to decrease hyperactive contractions in patients with diarrhea, Amitriptyline to decrease nerve pain and other issues in the GI system. Some people with Fabry also find that eating small meals, taking probiotics, and avoiding spicy, lactose-containing, or greasy foods also help decrease GI issues. There is an increased interest in the irritable bowel syndrome focused FODMAP diet for Fabry disease. Before beginning any new diet plan, please consult your primary care physician.
Dr. Claire Zar-Kessler at Massachusetts General Hospital is also enrolling in a study to learn more about gastrointestinal issues in Fabry disease. In order to find a Fabry disease specialist who can give you more information on treatment for GI symptoms in patients with Fabry disease, please refer to the National Fabry Disease Foundation online resource.
What can I expect during home-care infusions for Fabry disease?
The following content is sponsored by Optum Specialty and infusion Pharmacies
Nurses performing enzyme replacement therapy (ERT) home infusions for Fabry disease will follow the same treatment protocols as in a physician’s office or infusion clinic. The nurse will monitor vital signs, observe the patient during and after their infusion, and manage any side effects.
Although home infusions follow the same treatment protocols as facility based infusions, they are more personalized because the patient has one-on-one attention from the nurse. In addition, home-nursing visits can be scheduled based on patient and family availability.
If you or your family have questions about any differences between home infusions and infusions in a clinic, you should always feel free to ask your home infusion nurse or your doctor. In addition, some specialty infusion pharmacies, such as Optum Specialty and Infusion Pharmacies, have disease-specific advocacy teams who will walk you through the infusion process step-by-step and answer your questions along the way.
What can be done to help prevent Fabry disease from damaging my kidneys?
It’s important to understand that once kidney damage is done to the podocyte (filtering cells of the kidney) and other kidney cell types, it’s often difficult, if not impossible to reverse the damage. This is why it’s particularly important to work with your Fabry specialist and if they recommend it, visit a kidney doctor (nephrologist) at least once a year. They are the experts most qualified to discuss medications, kidney function, and treatment options with you.
Enzyme replacement therapy (ERT) and chaperone therapy are the only therapies for Fabry disease that address the underlying problem of glycolipid accumulation in the kidney. Many people with Fabry disease will need to combine ERT with another medications, called an angiotensin converting enzyme inhibitors (ACE inhibitor) and angiotensin receptor blockers (ARBs), to reduce the amount of protein in the urine due to kidney filtering problems and to best keep the kidneys working. A nephrologist experienced with Fabry disease can help manage these medications. The National Fabry Disease Foundation has a Fabry specialist finder on their website that can help you find a kidney doctor specializing in Fabry disease and the kidneys.
What are the symptoms of Fabry disease?
Fabry disease is often divided into two types, "classic" and "non-classic or late onset". Both forms lead to serious medical problems, but most often people with classic Fabry disease have earlier and more severe symptoms. Classic and non-classic Fabry disease affects both women and men, although symptoms vary more in women due to the way Fabry disease is passed through families.
In Classic Fabry disease, the early signs usually begin in childhood and include heat intolerance, sweating abnormalities, overheating while exercising, protein in the urine, a characteristic starburst pattern on the cornea seen during eye exams, a reddish-purple skin rash or lesions, burning or tingling pains in the hands and feet, chronic tiredness, and gastrointestinal issues, such as chronic diarrhea, bloating, and constipation. Usually the first signs of Fabry disease in kids are a tingling or pain in the hands and feet when overheated or sick and frequent bloating and diarrhea. In adults, these symptoms worsen and can also include decreasing kidney function, abnormal heart rhythms, severe abdominal pain, difficulty eating normal sized meals, numbness in the hands and feet, hearing loss, mini-strokes, fuzzy thinking, dizziness/vertigo, depression, anxiety, and panic attacks. Since the condition is progressive, untreated Fabry disease can lead to severe health problems such as kidney failure, heart disease, as well as nerve and brain problems, such as stroke. Not every person with Fabry disease will present with the same symptoms; however, without treatment, the disease will worsen over time. Some people with non-classic Fabry disease typically do not have childhood symptoms, but can have many of the same health problems as people with classic Fabry disease at a later age. Kidney and heart disease may occur much earlier than in the average person, so monitoring for Fabry-related health problems should start at the time of diagnosis.
To learn more about the health issues seen in Fabry disease, visit the Fabry Support and Information Group (FSIG) and the National Fabry Disease Foundation (NFDF).
There is also an excellent summary about Fabry disease written for doctors at Fabry Disease- GeneReviews.
What are the main symptoms of Fabry disease in children?
In classic Fabry disease, the early signs usually begin in childhood. Symptoms include tingling or burning pains in the hands and feet, sweating abnormalities, heat intolerance, protein in the urine, reddish-purple skin lesions called angiokeratomas, chronic tiredness, and gastrointestinal issue, such as chronic diarrhea, gas, and constipation. Usually the first signs of Fabry disease in kids are a tingling pain in the hands and feet when overheated or sick, frequent bloating, and diarrhea. Children with non-classic or late onset Fabry disease may have some or all of these symptoms and their health should be monitored in either type. Based on United States expert treatment recommendations and published guidelines from the European Fabry Working Group, patients of either sex with certain symptoms due to Fabry disease should be considered for treatment. Based on published cases of Fabry disease in children we have learned the following information about the possible timing of Fabry related health problems:
Ages 1 to 6:
Some children will have mild or no symptoms throughout their childhood, because the disease can vary in its severity and symptom presentation. As early as age 2, children with classic Fabry disease may have burning or tingling pain in their hands or feet when overheated. Doctors and medical professionals may refer to this pain as acroparethesias. Children may also experience Fabry "pain crises" that consist of episodes of severe pain lasting from a few minutes to several days, usually triggered by illness, fever, stressful conditions, or heat. As children as young as 3 ½ often cannot sweat, they have problems in hot conditions, while working, or during active play, as their body cannot cool correctly. Young children can begin having gastrointestinal problems, including diarrhea, nausea, bloating, abdominal pain, and vomiting as early as 1 year of age if they are affected by classic Fabry disease. They may begin to develop angiokeratomas, small, purple-red bumps that can be mistaken for a rash as early as age 4 years.
Ages 7 to 10:
As they progress through their childhood, many of the previously mentioned symptoms may worsen or become more obvious, like the angiokeratomas and gastrointestinal problems. On heart testing, children with Fabry may have a slow heartbeat (bradycardia) that doesn’t appear to cause any symptoms. Some children will have undetectable symptoms in their early childhood and begin to develop obvious problems after age 10.
Ages 11 to 18:
Although young teens tend to have similar symptoms as children, older adolescents and young adults may, in addition, begin developing kidney disease (e.g. proteinuria and worsening kidney function), heart palpitations/chest pain, and an enlarged heart. Evidence of these effects include protein in the urine, more severe pain crisis, heart palpitations, panic attacks, and shortness of breath. Many patients will have have an eye finding called corneal verticillata or more commonly described as a "propeller whorl". Most optometrists can diagnosis this eye finding with a slit lamp examination.
As the symptoms of Fabry disease progress, the quality of life can diminish and living with Fabry disease can cause depression (~46% of patients) and severe clinical depression (~28%). It is important to seek an evaluation for depression.
The best place to learn more about the early signs of Fabry disease in children, is a Fabry center. Find a Fabry center nearby at the National Fabry Disease online resource.
What are the gastrointestinal issues seen in Fabry disease?
Gastrointestinal issues (GI) in Fabry disease are different from person to person but often include: alternating cycles of chronic diarrhea and chronic constipation, stomach cramping and pain, bloating, frequent gas, nausea, vomiting, and feeling full early or early satiety. Studies have found that 52-60% of patients with Fabry disease have GI symptoms. GI issues can start as early as one year of age in children with Fabry disease. Many people with Fabry disease are also incorrectly diagnosed with Crohn’s disease, celiac disease, or irritable bowel syndrome (IBS). The GI symptoms in Fabry are thought to be caused by the storage of globotriaosylceramide (GL-3) in the body that interferes with nerve and cell function in the GI system. This disrupts how fast the stomach empties, how quickly food moves through the intestine, and other functions of digestion including absorbing nutrients. Treatment with enzyme replacement therapy [Fabrazyme (agalsidase beta) or Replagal (agalsidase alfa)] or chaperone therapy [Galafold (migalastat)] has been shown to help decrease the severity and frequency of GI symptoms. After an evaluation by gastroenterologist and the ruling out other non-Fabry related causes of GI issues, doctors may also try symptom-specific medications such as: Reglan (metoclopramide) to help the stomach empty, Zofran (Ondansetron) to reduce nausea and vomiting, pancreatic enzymes to aid digestion, Loperamide (Imodium) to decrease hyperactive contractions in patients with diarrhea, or Amitriptyline to decrease nerve pain and other issues in the GI system. Some individuals with Fabry also find that eating small meals, taking probiotics, and avoiding spicy, lactose-containing, or greasy foods also help decrease GI issues.
Dr. Claire Zar-Kessler at Massachusetts General Hospital is also enrolling in a study to learn more about gastrointestinal issues in Fabry disease.
What are the first steps after an initial diagnosis of Fabry disease?
After an initial diagnosis of Fabry disease, doctors will take a comprehensive medical history to look for signs and symptoms of disease and will also run tests to check kidney function, heart health, and neurological function. Individuals may be referred to a dermatologist for skin examination, to an ophthalmologist for an eye exam, to a cardiologist for check on heart function, to a audiologist for hearing assessment, and to medical genetics and a genetic counselor for a genetics consultation.
The best way to obtain the most accurate, current, clear, and comprehensive information is to be seen at a Fabry focused center that specialize in the treatment of patients with Fabry disease. At most centers you will see a Fabry-focused doctor, genetic counselor, and nurse who work as a team to answer your questions, discuss testing, identify your at-risk family members, and develop a comprehensive evaluation and treatment plan for you. The team will work with your current doctors to organize the treatment, tests, and specialists you need.
In order to find a Fabry disease specialist who can provide more information on Fabry disease, please refer to the National Fabry Disease Foundation online resource. Please also feel free to call the Emory Fabry Center at 800-200-1524 to locate a center in your state.
In the United Kingdom, there are certain centers that specialize in genetic disorders, such as Fabry disease. Speak to your physician about finding a specialist. Other parts of the world also have great resources and centers of excellence for Fabry disease. Again, talking to your doctor can be an important first step in finding a specialist.
What are the advantages of home infusion for Fabry disease?
The following content is sponsored by Optum Specialty and Infusion Pharmacies
For people who have Fabry disease, home infusions of enzyme replacement therapy (ERT) can be a safe and practical way to reduce the burden of treatment. The advantages of home infusion may include:
What are possible Fabry ERT infusion related problems or reactions I should watch for?
Sometimes during an infusion, people can have the following health issues:
Feeling Cold
Headache
Fever
Breathlessness
Chest Pain
Chills
Nausea
Itching
Racing Heart
Hives or bumps
What are additional medications that can help my kidney function in Fabry disease?
In addition to enzyme replacement therapy (ERT) or chaperone therapy, individuals with proteinuria and/or high blood pressure may be prescribed blood pressure lowering medicines called angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs). These medications have been found to protect the kidneys and decrease the amount of protein in the urine. The National Heart, Lung, and Blood Institute recommends that people with diabetes or reduced kidney function should keep their blood pressure below 130/80 mmHg. For more information about maintaining kidney health with Fabry disease, please talk to your Fabry doctor or genetic counselor.
Should I start treatment before I have Fabry symptoms?
In Fabry disease, globotriaosylceramide or GL-3 and related glycolipids start building up and are stored in the body’s cells before birth. Over time, serious organ damage can occur silently before an individual actually feels ill. For example, kidney biopsy studies have shown that there can be kidney damage before protein or other kidney function markers in the urine even appear.
In order to prevent Fabry disease from causing irreversible damage, medical evaluation and treatment need to occur as soon as possible. Early intervention with enzyme replacement therapy (ERT) or chaperone therapy offers the best protection against the complications and health problems related to Fabry disease. In adults, this means beginning ERT or oral therapy as soon as Fabry disease is diagnosed. In children with Fabry disease, the decision to begin therapy is based on the symptoms that they have and discussions of the risks and benefits of ERT with your Fabry disease specialist. There are also certain age restrictions for many of the available treatments. For more information about Fabry disease treatment and ERT, please talk to your Fabry doctor or refer to the National Fabry Disease online resource.
Should I change my diet if I have Fabry disease?
Individuals living with Fabry disease with normally functioning kidneys do not typically need to change their diet. People with reduced kidney function need to be aware that some parts of a normal diet may speed their kidney failure. Dietitians often work with kidney doctors to help patients with kidney disease limit the amount of protein, cholesterol, potassium, and/or sodium they eat so that the kidneys have less work to do. Some people with Fabry and normal kidney function find that eating small meals. taking probiotics, and avoiding spicy, lactose-containing, or greasy foods also help decrease GI issues.
Should I be tested for Fabry disease?
The symptoms that occur in Fabry disease can also be found in other diseases and conditions. Your doctor can help you decide if you should be tested for Fabry disease and refer you to a specialist for additional guidance.
Before testing, it is also important to think about how you would feel if the test results are positive for Fabry disease. What would it mean for your family members? Since Fabry is passed through families, there would be a chance that other family members may also have Fabry disease. What would evaluation and treatment be like? Please talk to your doctor or a genetic counselor to discuss the implications for both yourself and your family before testing. Take your time when deciding to get tested for Fabry disease.
A genetic counselor can also help you think about the issues surrounding testing for Fabry disease. Genetic counselors can be found on the [link url=" https://www.findageneticcounselor.com/” target=”_blank”>National Society of Genetic Counselors website.
Is there newborn screening for Fabry disease?
In the United States, there is newborn screening for Fabry disease in a few states. These states do testing for enzyme levels of alpha-galactosidase A, or alpha-Gal, on newborn dried blood spots. Currently, Missouri, Illinois, Oregon, Tennessee, and Maryland screen all babies born in the state for Fabry disease. Pennsylvania and New York offer the option for Fabry disease testing on the newborn screening (in selected areas of New York City). New Jersey recently began screening for Fabry disease and New Mexico will soon offer newborn screening. Not all states test for Fabry disease as it has not yet been added to the Recommended Uniform Screening Panel (RUSP) that many states use to decide which new tests to begin adding to their newborn testing. A current listing of states performing newborn screening for Fabry disease can be found on the NewSTEPs website in the data visualizations section.
It is important to remember that a "positive" newborn screen for Fabry disease does not mean that a child has the condition. A false positive can occur for many different reasons and there are important follow up tests that need to be done to determine if a child actually has Fabry disease.
For more information about newborn screening for Fabry Disease, please refer to the Baby’s First Test online resource.
In order to find a Fabry disease specialist who can provide more information on newborn screening for Fabry disease, please refer to the Fabry specialist finder online resource.
Is there gene therapy for Fabry disease?
Fabry is believed to be a good condition to treat with gene therapy because it is caused by a single gene (GLA) and it doesn’t take a large amount of working enzyme to make a difference in the medical issues. For gene therapy to work, scientists need to figure out the best way to get the working gene into the right place in the DNA in as many cells as possible WITHOUT causing side effects like an allergic reaction.
Although not yet available to everyone, gene therapy studies in Fabry disease are open to enrolling people with Fabry disease in the United States, Canada, Australia, and other countries. Most of the studies are focused on men with classic Fabry disease at this time, but in future studies women with classic Fabry disease and individuals living with non-classic Fabry disease will be included as well.
Currently, there are several companies (4DMT, Freeline, Sangamo) exploring different methods of doing gene therapy in Fabry disease. While the gene therapy is still in various clinical and preclinical phases of development, it is important to discuss the risk and benefits of participating in clinical trials with new biological agents with a physician familiar with the options.
The easiest way to learn if you are a candidate for a gene therapy trial in Fabry disease is to talk to a healthcare provider that specializes in Fabry disease. However, studies focusing on gene therapy are also posted at clinicaltrials.gov where you can search for "Gene Therapy Fabry disease." It may help to avoid limiting your search to your state or country, as many studies will pay for your travel if you are eligible to join.
Is there any research demonstrating an association between Fabry disease and an enlarged nuchal translucency?
There are no formal studies that give us much information about the relationship between nuchal translucency in pregnancy and Fabry disease. A nuchal translucency (NT) is a fluid-filled area at the back of a baby’s neck seen on ultrasound between 11-14 weeks of pregnancy. A NT is great indicator of the health of the baby and pregnancy. A large or enlarged NT can be a sign that the baby has a heart problem, a separate genetic condition, or a chromosome abnormality caused by having missing or extra chromosomes, such as Down syndrome or Turner syndrome. Some babies with a large NTs early in pregnancy are completely fine at birth with no health problems. Prenatal genetic counselors are available to talk to patients about the risks associated with a large NT and help them make decisions about testing. At this point, doctors believe a large NT in pregnancy is unlikely to be related to Fabry disease.
Is there a treatment for Fabry disease?
Currently there are two medications approved by the Federal Food and Drug Administration (FDA) for treatment of Fabry disease in the United States: an enzyme replacement therapy (ERT) and a chaperone therapy.
The FDA approved enzyme replacement therapy (ERT) treatment for Fabry disease in the United States is called Fabrazyme®, or agalsidase beta, and is made by a company called Sanofi Genzyme Corporation. The goal of ERT is to replace the enzyme missing in individuals with Fabry disease, with artificial or recombinant alpha-Galactosidase A or alpha-Gal, so that their body can breakdown stored glycolipids and prevent them from being stored in cells within the body. Patients getting ERT are infused every other week and receive the enzyme by an intravenous (IV) infusion. When began early in the course of the disease, replacing the enzyme helps slow the progression of the disease, reduces complications, and may even prevent long-term complications. Fabrazyme® is a treatment that can be given to individuals with any mutation or change in the GLA gene.
The FDA approved chaperone therapy for Fabry disease in the United States is called Galafold®, or migalastat, and it made by a company called Amicus Therapeutics. Galafold® is pill taken every other day that is designed to help an individual with Fabry disease’s own enzyme work more effectively (chaperone therapy). Galafold® only works for specific mutations in the GLA gene that are considered "amenable". Individuals with Fabry disease can talk to their doctors to learn if they may be able to take Galafold and check to see if their GLA change is considered at the GLA mutation search website.
Along with ERT and chaperone therapy, there are also treatments available to help relieve some of the other symptoms associated with Fabry disease. Drugs such as diphenylhydantoin, carbamazepine and gabapentin have been helpful in reducing nerve pain and tingling in the extremities of individuals with Fabry disease. Individuals with proteinuria and/or high blood pressure may be prescribed blood pressure lowering medicines called angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs). These medications have been found to protect the kidneys and decrease the amount of protein in the urine. Lastly, to help relieve some of the gastrointestinal discomfort, medications such as Reglan (metoclopramide) can be used to help the stomach empty, Zofran (ondansetron) to reduce nausea and vomiting, pancreatic enzymes to aid digestion, and Imodium (loperamide) to decrease hyperactive contractions in patients with diarrhea. Some people with Fabry also find that eating small meals, taking probiotics, and avoiding spicy, lactose-containing, or greasy foods also help decrease GI issues.
For more information about Fabry disease treatment please talk to your Fabry doctor or find a Fabry center at the National Fabry Disease online resource.
Is my lipid panel abnormal because I have Fabry disease?
The compounds that build up in the body with Fabry disease are called globotriaosylceramide (GL-3) and lyso-globotriaosylceramide (lyso-GL3) . Lyso-GL3 and GL-3 are not the same as cholesterol or triglycerides and are not impacted by the food people eat. Having high, harmful "bad" LDL cholesterol or high triglycerides is not part of Fabry disease. However, whether or not someone has Fabry disease, having high triglycerides or LDL cholesterol does increase the risk for a person to have heart problems. So, even though Fabry disease doesn’t make it more likely for a person to have high LDL cholesterol issues or dyslipidemia, everyone with Fabry disease should have their cholesterol levels checked so that if it is high they can be treated by their doctor.
For additional questions about these lipid monitoring tests and when they should be scheduled, please refer to a Fabry doctor or genetic counselor. In order to find a Fabry disease specialist or more information on Fabry disease, please refer to the National Fabry Disease Foundation online resource.
Is it safe to treat Fabry disease in the home setting?
The following content is sponsored by Optum Specialty and Infusion Pharmacies
Studies have shown that home infusion of enzyme replacement therapy (ERT) is a safe and effective alternative for many patients living with Fabry disease. Before beginning home infusions, patients should be stable – meaning they’re not having active reactions to infusions. It’s also important to make sure home infusion nurses are properly trained on mixing ERT medications, infusing ERT, and managing possible infusion-associated reactions.
The manufacturers of some ERT medications have developed home infusion guides to help with the infusion experience. In addition, some pharmacies experienced with home infusion of ERT, such as Optum specialty and infusion pharmacies, have worked with treatment centers and doctors to further develop training and infusion protocols to simplify the complex treatment of Fabry disease.
Is Fabry disease found more frequently anywhere in the world?
Fabry disease (FD) is found in individuals of all different backgrounds and ethnicities around the world; however, there are a few places where it is seen more frequently. For example, there is an increased number of people in Nova Scotia, Canada with classic Fabry disease and in Taiwan with nonclassic Fabry disease.
Is ERT beneficial in Fabry individuals after a kidney transplant?
Small studies of people with Fabry disease after a kidney transplant indicate that enzyme replacement therapy (ERT) is safe and works against the other non-kidney related problems of Fabry disease. In one small study of three kidney transplant patients treated with ERT at a dose of 1 mg/kg every 2 weeks for 13-18 months, the blood plasma GL-3 levels decreased (in all 3 patients), hand/foot/arm/leg pain went away in the one patient experiencing pain, and heart problems were stabilized or improved in 2 out of 3 transplant patients studied. It’s important to remember that while the transplanted kidney will have high levels of alpha-galactosidase A to keep the transplanted kidney free of Fabry disease, it typically is not enough enzyme to prevent further Fabry symptoms all over the body. Based on this information, many doctors will recommend staying on ERT to help prevent other Fabry related health problems and events from occurring after kidney transplant. For more information about Fabry disease treatment and ERT after kidney transplant, please talk to your Fabry doctor or genetic counselor.
If you know the family mutation of Fabry, do you then know what organs will most likely be affected?
There are many, many family specific mutations in GLA that cause Fabry disease. If a male has a "classic" gene mutation, then doctors have a better idea which organs will be affected and a general time frame of problems such as kidney disease. If a woman has a "classic" gene mutation in GLA, then it gets more complicated. In this case we know that at least 75% of women will have issues in their heart, kidneys, brain, digestive tract, eyes and/or nerves, but it is not a guarantee. Additionally, if an individual with Fabry disease is on Fabry specific therapy such as Fabrazyme or Galafold, then they will change the course of the disease (hopefully for the better).
In most people (male and female) with "nonclassic" or "later-onset" mutations in the GLA gene, Fabry specialists don’t really know which organs will be affected and when. Doctors will research the gene change and work with each person as an individual and watch for the usual organs for complications and review family history to provide the best care and predictions possible.
It gets complicated, but doctors are trained to perform the tests that watch each organ that could be affected by Fabry and and tailor the treatment to the individual. The National Fabry Disease Foundation has a Fabry specialist finder on their website that can direct individuals with Fabry disease to Fabry specialists who can help predict health issues with specific GLA gene mutations.
If I have Fabry disease, do I have to stop smoking?
Smoking increases the risk for kidney failure, strokes, heart attacks, lung cancer, and respiratory illness. Since individuals with Fabry disease are already at increased risk for stroke, heart attack, kidney failure, and lung problems, it is recommended to stop smoking. Individuals with Fabry disease can talk to their doctor about safe strategies for quitting smoking.
I have Fabry disease. Will my children have it?
Fabry disease is inherited in an X-linked pattern. Both men and women can experience symptoms of the disease.
The chance for a child to inherit Fabry disease depends on whether the father or the mother has Fabry disease. If a man has Fabry disease, then his only X-chromosome contains the nonworking GLA gene and he will pass it on to all of his daughters and none of his sons. If a woman has Fabry disease, only one of her X-chromosomes contains the nonworking GLA gene. This means she will have a 50% chance with each pregnancy of passing it on to both her daughters and sons. Both males and females can have Fabry disease.
If a child is the first in the family to be diagnosed with Fabry disease, it was likely inherited from one of their parents who has not yet been diagnosed, although spontaneous (de novo) gene changes that occur for the first time in that child are possible as well. Each parent of a child with Fabry disease should be tested for the same known mutation in the GLA gene.
If Fabry disease runs in a family, genetic testing is available to determine who else in the family is affected. Free testing options are available through Discover Fabry. For more information about testing, you should speak with your healthcare provider. A genetic counselor can also help determine an individual’s risk of having Fabry disease based on a detailed family and medical history as well as discuss and organize testing. For individuals who live in the United States, to find a genetic counselor in your area use the National Society of Genetic Counselors Find a Genetic Counselor tool on the National Society of Genetic Counselors website.
I have Fabry disease. How is depression treated?
Patients with Fabry disease may experience psychological symptoms common to other chronic illnesses, including depression and anxiety. Appropriate treatment can help most people who suffer from depression. Current treatments include medications and talk therapies with therapists that ease the impact of depression. The first step in determining a treatment path is a psychological evaluation by a psychiatrist and/or a psychologist. A diagnostic evaluation will include a complete history of symptoms such as: when they started, how long they have lasted, how severe they are, whether the patient had them before and, if so, whether the symptoms were treated and what treatment was given. In individuals affected by Fabry disease, studies have shown that treatment with enzyme replacement therapy (ERT) does improve quality of life, as well as physical health which may help with overall life outlook. Some Fabry centers have psychologists skilled in evaluating or treating Fabry-related depression, anxiety, and panic attacks. Additional information about support groups and strategies for coping with the day-to-day challenges of living with Fabry disease can be found on the Discover Fabry Support online resource.
I have Fabry disease. How do I know if I am depressed?
Everybody has times when they feel sad or low. However, the "blues" can be more severe in some individuals and last longer than usual. This is called depression. Depression is an illness that involves the body, mood, and thoughts. It affects the way a person eats and sleeps, the way one feels about oneself, and the way one thinks about things. Depression is not a sign of personal weakness or a condition that can be willed or wished away. People with a depressive illness cannot merely "pull themselves together" and get better. Depression often interferes with normal functioning and causes pain and suffering not only to those who have a disorder, but also to those who care about them. Signs that suggest depression include:
I have Fabry disease. How are panic attacks treated?
Patients with Fabry disease may experience psychological symptoms common to other chronic illnesses, including panic attacks. Effective treatments for panic disorders are available in the form of medications and psychotherapy. These treatments are similar to those used to treat depression and anxiety. In individuals affected by Fabry disease, studies have shown that treatment with ERT does improve emotional as well as physical health. Some Fabry centers have psychologists skilled in evaluating or treating Fabry related depression, anxiety, and panic attacks. Additional information about support groups and strategies for coping with the day-to-day challenges of living with Fabry disease can be found on the Discover Fabry Support online resource.
I have Fabry disease; are home infusions right for me?
The following content is sponsored by Optum Specialty and Infusion Pharmacies.
Home infusions can be a convenient option for many patients living with Fabry disease and receiving enzyme replacement therapy (ERT). When starting ERT, most doctors prefer that patients begin infusions in a hospital or outpatient infusion center. After a period of time – determined by the ordering doctor (usually three to six months) – patients may have the option to transition to home infusions. Patients might be a candidate for home infusion if:
I am living with Fabry disease. What are panic attacks?
Panic attacks are repeated episodes of intense fear that strike often and without warning. Symptoms of panic attacks include any of the anxiety symptoms listed above, as well as the following:
I am living with Fabry disease. What signs indicate that I am anxious?
Many people feel anxious before exams, presentations, or first dates. Individuals affected by anxiety disorders have constant, unremitting worry and fears about everyday activities. Signs and symptoms of an anxiety disorder include:
How soon after starting enzyme replacement therapy for Fabry disease can I transition to home care?
The following content is sponsored by Optum Specialty and Infusion Pharmacies.
Most doctors prefer that people who have Fabry disease begin enzyme replacement therapy (ERT) infusions in a hospital or outpatient infusion center. After a period of time – determined by the ordering doctor (usually three to six months or seven to 12 infusions) – many patients have the option to move to home infusions for their therapy.
The waiting period before moving to a home setting for infusions is to make sure the patients are comfortable with infusions and the doctors have time to manage any infusion-associated reactions (IARs) that may occur. Not all people receiving ERT will have IARS, but in clinical trials with Fabrazyme, 59% of patients reported IARs, some of which were severe. Doctors will be looking for signs of IARs so that they can manage them prior to referring patients to home infusions.
How should gastrointestinal symptoms in Fabry disease be monitored?
Doctors who focus on gastrointestinal (GI) issues are called gastroenterologists or GI doctors. Fabry focused healthcare providers and GI doctors work together to learn more about a specific person’s GI issues and come up with the best treatment plan. Important things to know when putting together a treatment plan include:
How safe is ERT for Fabry disease?
Multiple research studies have looked at the safety and side effects of enzyme replacement therapy and found it to be safe for use in humans. In the studies, the people with Fabry disease given ERT had fewer serious Fabry-related health problems than people with Fabry disease that were not on ERT. While ERT is generally safe and well tolerated, individuals treated with ERT can have infusion-related side effects or reactions. These infusion reactions are usually not life threatening and are most often fixed by slowing the speed of the infusion and giving medications before the infusion starts. The most commonly occurring side effects were fever and shaking (rigors) during the infusions.
Rarely, severe reactions can occur in patients during ERT infusions. Approximately 1% of patients developed anaphylactic or severe allergic reactions. If severe reactions occur, the doctor or nurse will immediately stop the administration of ERT and provide emergency treatment.
For more information about Fabry disease treatment and ERT, please talk to your Fabry doctor or refer to the National Fabry Disease online resource.
How many people in the US have Fabry?
Around 5,000 people in the U.S. have been diagnosed with Fabry disease. That number is a mixture of men and women living with Fabry disease, as well as a combination of people with classic and non-classic Fabry disease. However, doctors fully believe that many people living with Fabry disease have not been diagnosed due to the nonspecific health problems associated with the condition (e.g. severe tiredness, burning pain in the hands and feet). Newborn screening has increased the number of people diagnosed with Fabry disease, but it has also made things a little more complicated. Some newborns and their family members have new changes or variants of unknown significance in the GLA gene and it is unclear if these changes contribute to the health issues associated with Fabry disease.
How many companies are conducting gene therapy studies in Fabry disease?
Gene therapy studies in Fabry disease are open to enrolling people with Fabry disease in the United States, Canada, Australia, and other countries. Most of the studies are focused on men with classic Fabry disease at this time, but in future studies women with classic Fabry disease and individuals living with non-classic Fabry disease will be included as well. There are several companies (4DMT, Freeline, Sangamo) exploring different methods of doing gene therapy in Fabry disease, so it is important to discuss the risks and benefits with a doctor familiar with the options to see which appears to make the most sense for a specific individual.
The easiest way to learn if you are candidate for a gene therapy trial in Fabry disease is to talk to healthcare provider that specializes in Fabry disease. However, studies focusing on gene therapy are also posted at clinicaltrials.gov where you can search for "Gene Therapy Fabry disease." It may help to avoid limiting your search to your state or country, as many studies will pay for your travel if you are eligible to join.
How long do you have to stay on ERT for Fabry disease?
ERT is a life-long treatment. As soon as ERT is stopped, the GL-3 begins storing up again. When this happens, the Fabry symptoms and damage will progress again. ERT is an effective treatment, however it constantly requires infusions every two weeks. Research is currently being conducted to find a longer-lasting treatment. For more information about Fabry disease treatment and the importance of staying consistent with ERT, please talk to your Fabry doctor or refer to the National Fabry Disease online resource.
How is kidney function measured?
Doctors monitor kidney function in people living with Fabry disease to watch for signs of kidney disease. These are important tests as damage to the kidneys could occur without the individual even knowing it. Kidney function tests may include:
How is Fabry disease inherited?
Fabry disease is passed through families in an X-linked inheritance pattern, meaning the GLA gene that causes Fabry disease is located on the X-chromosome. Women have two copies of the X-chromosome (XX) and men have one copy of the X-chromosome and one copy of a Y-chromosome (XY). If men inherit an X chromosome containing the non-working GLA gene, they are unable to produce alpha-Gal enzyme. Without alpha-Gal, GL-3 builds up in the body and causes the symptoms and health problems of Fabry disease. On the other hand, if women inherit an X chromosome with a nonworking GLA gene, they have a second X-chromosome with a working GLA gene that can produce some alpha-Gal enzyme. In the past, it was believed that women who were "carriers" would not have Fabry related health problems because they had a normal second copy of the gene. However, women are not just carriers and they can have Fabry related health problems. Fabry disease is unique, as most X-linked disorders affect males primarily. It is important to speak to your physician about all your signs and symptoms and not rule out Fabry disease if you are a female.
The chance for children to have Fabry disease depends on whether the father or the mother has Fabry disease and the X-chromosome with the non-working GLA gene. If a man’s X-chromosome contains the nonworking GLA gene, he will pass it on to all of his daughters and none of his sons. If one of a woman’s X-chromosomes contains the nonworking GLA gene, she will have a 50% chance with each pregnancy of passing it on to both her daughters and sons. Both males and females can have Fabry disease.
To find a genetic counselor in your area who can discuss the chance for someone with Fabry disease to have a child with Fabry disease or discuss genetic testing options, use the National Society of Genetic Counselors Find a Genetic Counselor tool on the National Society of Genetic Counselors website.
How is anxiety treated in people with Fabry disease?
Patients with Fabry disease may experience psychological symptoms common to other chronic illnesses, including anxiety, panic attacks, and depression. Appropriate treatment can help most people who suffer from anxiety. Current treatments include medications and talk therapies with a psychologist. A number of medications that were initially approved for treating depression have been found to be effective for anxiety disorders as well. If one medication is not effective, others can be tried. In individuals affected by Fabry disease, studies have shown that treatment with enzyme replacement therapy (ERT) does improve emotional as well as physical health. Some Fabry centers have psychologists skilled in treating Fabry related depression, anxiety, and panic attacks. Additional information about support groups and strategies for coping with the day-to-day challenges of living with Fabry disease can be found on the Discover Fabry Support online resource.
How effective is ERT for treating Fabry disease?
Enzyme replacement therapy (ERT) can help stabilize, delay, or prevent the progression of serious health problems if started early in the disease’s course. The research studies have shown that ERT reduces the amount of stored glycolipids, specifically globotriaosylceramide or GL-3 in urine, blood, kidney cells, skin cells, and heart cells.
One study with Fabrazyme showed that at 11 months of treatment every other week, 94-96% of patients had no GL-3 in kidney cells on kidney biopsy, 85% of patients’ heart cells (myocytes) were clear of GL-3, and all of the patients’ skin cells from skin biopsy were clear of GL-3. Over the course of 35 months, patients on ERT were 61% less likely to have a serious Fabry disease event (kidney failure, heart attack, stroke, or death) than untreated patients. Treatment works best in patients treated early in their disease as children or teenagers. Similar data is available for ERT for Agalsidase alfa (Replagal™), another Fabry ERT medication that is not approved for use in the United States by the FDA.
When the amount of GL-3 is reduced in the body, the disease progression should slow. However, depending on the stage of Fabry disease, irreversible damage or scarring may have already been done to some organs. For example, if an individual already has serious kidney disease, ERT cannot stop the kidney failure, but can hopefully increase the time until dialysis/transplant is needed, and reduce the chance of heart disease and strokes. In addition, for an individual who had strokes, the hope is that the ERT will help prevent future strokes, heart attacks, and kidney disease. More research into the effectiveness of ERT and damaged organs is still needed.
It is important that a diagnosis is made before irreversible damage has occurred, as the treatment works best in patients treated early in their disease course.
For more information about Fabry disease treatment and ERT, please talk to a Fabry focused doctor who can be found on the National Fabry Disease online resource.
How does migalastat help Fabry disease?
Migalastat or Galafold® is a chaperone therapy pill taken every other day to treat Fabry disease. Migalastat is designed to work on the body’s natural alpha-galactosidase A enzyme by helping make the misfolded enzyme more stable and chaperoning it to the lysosome to do its job. The enzyme should then be able to break down stored glycolipids and prevent them from being stored in cells within the body. Based on studies that look at the gene changes or disease-causing mutations in people with Fabry disease, about 60% of individuals with Fabry disease have amenable mutations, which means they are responsive to treatment with migalastat and the drug may be an option for them to treat Fabry disease. Individuals with Fabry disease can talk to their doctors to learn if they may be able to take migalastat and check to see if their GLA change is considered amenable at the GLA mutation search website.
How does Fabry disease affect the lungs?
Much like other body systems, Fabry disease can also affect the lungs. This means it is best for people with Fabry disease to avoid smoking cigarettes, pipes, or other inhaled tobacco products. To watch for any lung problems, it is important to have pulmonary function tests every couple of years and be treated if any signs of breathing issues occur. Sometimes in adulthood, people with Fabry disease can have issues with shortness of breath that is more like chronic obstructive pulmonary disease than asthma. Of course, there are many causes of shortness of breath, so if you have Fabry disease and difficulty breathing it is good to have an evaluation with a lung doctor or pulmonologist. Please talk to your Fabry doctor or genetic counselor for more information about how to protect your lungs from Fabry disease.
How do I tell my children they have Fabry disease?
When you feel that your child is old enough and mature enough to understand the basics of the disease, you should set aside a time and a place for discussion. Be open to any questions that your child has and be honest about the answers. Ask them to tell you about Fabry Disease to make sure they understand and are involved in the conversation. You do not have to tell them everything in one discussion, since you can always add in more complicated details as your child ages. There are several children’s books about Fabry disease such as "Joe Learns About Fabry disease", "Dani Goes to Fabry Camp", "The Mystery of the Smashed Statue", and "The Long Road to Fabry" which are available on Amazon and may help your child understand Fabry disease better.
How do I talk to my doctor about home-infusion options for enzyme replacement therapy and Fabry disease?
The following content is sponsored by Optum Specialty and Infusion Pharmacies
Individuals living with Fabry disease who are interested in moving to home infusions should talk to their doctor about their interest in receiving their enzyme replacement therapy (ERT) at home. The doctor will decide if home infusions are a good option at that time, and if not, what would be required before the patient could begin ERT infusions at home.
Doctors will consider many factors, but they’ll usually make sure that at least these three things are true about the patient:
How do I know if my Fabry disease variant is amenable to Galafold?
Individuals with Fabry disease can talk to their doctors to learn if they may be able to take migalastat (Galafold®) and check to see if their GLA change is considered amenable at the GLA mutation search website. Migalastat is a chaperone therapy pill taken every other day to treat Fabry disease. Migalastat is designed to work on the body’s natural alpha-galactosidase A enzyme by helping make the misfolded enzyme more stable and chaperoning it to the lysosome to do its job. The enzyme should then be able to break down stored glycolipids and prevent them from being stored in cells within the body. Based on studies that look at the gene changes or disease-causing mutations in people with Fabry disease, about 60% of individuals with Fabry disease have "amenable mutations" and migalastat may be an option for them to treat their Fabry disease.
How do I find gene therapy studies for Fabry disease?
Gene therapy studies in Fabry disease are open to enrolling people with Fabry disease in the United States, Canada, Australia, and other countries. Most of the studies are focused on men with classic Fabry disease at this time, but in future studies women with classic Fabry disease and individuals living with non-classic Fabry disease will be included as well. There are several companies (4DMT, Freeline, Sangamo) exploring different methods of doing gene therapy in Fabry disease, so it is important to discuss the risks and benefits with a doctor familiar with the options to see which appears to make the most sense for a specific individual.
The easiest way to learn if you are candidate for a gene therapy trial in Fabry disease is to talk to healthcare provider that specializes in Fabry disease. However, studies focusing on gene therapy are also posted at clinicaltrials.gov where you can search for "Gene Therapy Fabry disease." It may help to avoid limiting your search to your state or country, as many studies will pay for your travel if you are eligible to join.
How do I explain Fabry fatigue to friends/family?
It can be very difficult for friends and family to understand the severe fatigue individuals with Fabry disease fight on a daily basis. A good way to explain the severe fatigue associated with Fabry disease is to say it feels like having the flu every day. It’s not just exhaustion, people with Fabry disease are sick and have to fight even to have the energy to stand up. It may also be helpful to explain that people living with Fabry disease have good days with higher energy levels and bad days where they are unable to leave the couch without a fight. Some people living with Fabry disease describe this as a roller coaster with fast ups and downs.
How common is Fabry disease?
Fabry disease (FD) is found in individuals of all different backgrounds, races, and ethnicities. Fabry disease occurs in a range of 1 in 1,250-117,000 live births worldwide.
How can I move to a different city and still get good Fabry care?
When an individual has Fabry disease, one of the most important things to learn about is how to get Fabry disease expert care in the new location. The easiest way to figure this out is to talk with their current Fabry expert and/or contact one of the Fabry support groups. Depending on an individual’s specific needs and symptoms, it may be crucial to figure out if there are good heart or kidney doctors in that new place that are comfortable working with patients with Fabry disease. Individuals on enzyme replacement therapy (ERT) will need to figure out if they can do home therapy or therapy in a clinic/hospital in the new location. It may be nice to reach out to other patients with Fabry in the new area to figure out how things would be different from their current care. A diagnosis of Fabry disease should not prevent a person from moving to a new place, but it does mean there is some research to find out ways to get the best care.
A list of treatment centers can be found online at The National Fabry Disease Foundation Fabry specialist finder website.
How can I get treated for Fabry disease if I am uninsured?
There are many resources to help you get the treatment you need for your Fabry disease (even without insurance). The usual process works like this:
Fabry disease has caused my kidneys to fail and I’m on dialysis now, how does this affect my treatment plan?
If an individual has Fabry disease and begins enzyme replacement therapy (ERT) with an elevated creatinine level or is already in end-stage renal disease, permanent damage to kidney cells has already occurred. In these cases, ERT or chaperone therapy cannot stop or prevent kidney failure, but can hopefully prolong and/or stabilize kidney function and reduce the chance for complications related to cardiac problems and strokes. Just as in individuals without Fabry disease, if your kidneys stop working correctly, you will need to undergo dialysis or kidney transplantation. If you choose to pursue dialysis, in order to remove the wastes from your body, dialysis must be attended exactly as prescribed by your nephrologist. For more information about maintaining kidney health with Fabry disease, please talk to your Fabry doctor or genetic counselor.
Even though I’ve been diagnosed with Fabry disease, I don’t feel sick, do I need to be evaluated?
Fabry disease is a progressive, life-threatening condition that can cause serious damage to the heart, kidneys and central nervous system if left untreated. Early signs and symptoms, such as pain and tingling in the extremities, heat intolerance, stomach issues and skin lesions, can vary from person to person. Regardless of whether or not an individual has very severe symptoms or no symptoms at all, globotriaosylceramide or GL-3 is stored in the body’s cells over time and serious organ damage can occur silently before an individual actually feels ill. For example, kidney biopsy studies have shown that there can be kidney damage before protein or other kidney function markers in the urine even appear. In order to stop or slow Fabry disease from causing irreversible damage, a medical evaluation and treatment need to occur as soon as possible. Early intervention with enzyme replacement therapy (ERT) or chaperone therapy offers the opportunity to stabilize many of the complications and health problems related to Fabry disease. In adults, this means beginning Fabry specific treatment as soon as Fabry disease is diagnosed. In children with Fabry disease, the decision to begin therapy is based on the symptoms they have and discussions of the risks and benefits of ERT with a Fabry disease specialist. In order to find a Fabry disease specialist or more information on Fabry disease treatment, please refer to the National Fabry Disease Foundation online resource.
Does insurance pay for home care for patients with Fabry disease?
The following content is sponsored by Optum Specialty and Infusion Pharmacies
Home infusions of enzyme replacement therapy (ERT) can be an excellent option to treat Fabry disease. In fact, many insurance companies allow – and some prefer – home infusion as an alternative to infusions in a hospital outpatient setting.
Once a doctor has decided if home infusions are appropriate for a specific patient, the next step is to see if the insurance plan allows home infusions. The drug manufacturer’s patient-service program and a specialty infusion pharmacy – such as Optum Specialty and Infusion Pharmacies – will check with the insurance company to help patients understand their options. Even if the primary insurance is Medicare, patients may be able to have home infusions. Certain Medicare Advantage plans and patient assistance programs work with Medicare Part D to provide full coverage.
Moving to home infusions is not something that patients and families have to figure out on their own. Drug manufacturer’s patient-service programs, doctors, and infusion providers such as Optum Specialty and Infusion Pharmacies, can help guide them through the insurance process and address issues as they appear.
Does Fabry disease cause itchy feet?
In Fabry disease, the build up of GL-3 and related lipids damages small nerve fibers and interferes with the normal functioning of nerve cells. Individuals often experience itching, numbness, tingling, burning, pain, sensitivity to cold, and problems sweating in their hands and feet due to the damage. Medical professionals and doctors may also refer to this as acroparesthesias or neuropathy. Enzyme replacement therapy (ERT) and/or prescription medications such as diphenylhydantoin, carbamazepine and gabapentin have been successful in some individuals living with Fabry disease in alleviating nerve pain.
There are many ways that individuals living with Fabry disease may describe the pain they feel in their hands and feet. Itchy feet, sand in the shoes, dragon fire in the feet and hands, frostbite, and other terms have all been used to describe this type of pain.
Do I still need to go to specialists and have monitoring tests when I am on ERT for Fabry disease?
Enzyme replacement therapy is only part of the treatment for Fabry disease. Patients on ERT will still need yearly monitoring tests to watch for any health problems and will also need to see specialist doctors to treat any problems that arise. Patients will need to continue taking any medications prescribed by their physicians.
For more information about Fabry disease treatment, please talk to a Fabry focused healthcare provider who can be found on the National Fabry Disease online resource.
Do I need to do anything special for my first Fabry ERT infusion?
It is important to drink plenty of water the night before and the morning of your infusion. Being well hydrated makes starting the IV for the infusion line much easier, so it’s important to be hydrated before all infusions. Note: if you are on dialysis or have another reason for careful fluid intake monitoring, continue to follow your doctor’s instructions. On the morning of the infusion, eat breakfast as normal and prepare for a long day. Please talk to your Fabry doctor or genetic counselor for more information about what to expect with ERT treatment and how to prepare for your first infusion.
Do I have to be tested for Fabry disease?
If your family history suggests that you are at risk to be affected by Fabry disease, but you decide not to have a genetic test, you can still pursue all the services (kidney testing, heart evaluations, brain imaging, etc) available to people with Fabry disease. A genetic test confirmation is not required in order to have screening. However, without a diagnosis, you won’t be able to accurately assess the risk of passing Fabry disease to your children, you will be unable to get treatment with the FDA approved medication for Fabry disease, and it may be difficult to have monitoring tests covered by your insurance. Please talk to your doctor or genetic counselor to discuss the implications for both yourself and your family before testing. Take your time when deciding to get tested for Fabry disease.
For more information about how to talk to your doctor about Fabry disease or how to get tested for Fabry, it may be helpful to reach out to a medical professional experienced in working with people with Fabry disease. A list of these Fabry centers can be found at the National Fabry Disease Foundation’s clinical center list.
To find a genetic professional near you who can discuss genetic testing issues and options, consider consulting a genetic counselor. Genetic counselors can be found on the National Society of Genetic Counselors website by using the Find a Genetic Counselor tool.
Can I switch from ERT to Galafold (migalastat) for Fabry disease treatment?
Individuals with Fabry disease who are 18 years of age or older and have Galafold (migalastat) "amenable mutations" can speak with their doctors to determine if switching from ERT to migalastat may be an option for them. If it is, the Fabry disease specialist will usually collect blood and urine to test biomarkers before therapy is changed and then monitor them again at specific time points after migalastat is begun. The plan for switching a specific patient from ERT to treatment with migalastat is something to ask a Fabry disease specialist. In order to find a Fabry disease specialist who can provide more information on Fabry disease, please refer to the National Fabry Disease Foundation online resource.
Can I keep the same home-care nurse to provide my Fabry infusions if I switch home-care companies?
The following content is sponsored by Optum Specialty and Infusion Pharmacies
When patients living with Fabry disease find a nurse who is excellent at providing their home infusions, it can be very frustrating when insurance or life events require a change in nursing services. Don’t despair! In many cases, home-health nurses often work with more than one home-health company. Talk to the new home-health company and the nurse to see what options you have.
For example, Optum Specialty and Infusion Pharmacies works closely with many qualified contractors and nursing agencies around the United States and, in many cases, they can train and certify preferred nurses to provide infusion services for the home infusion therapy. For questions, the patient or nurse can call the Diplomat Clinical Team at 888.335.4279.
Can I get on disability if I have Fabry disease?
Some people living with classic Fabry disease have life impacting symptoms, do not work, and are on disability. Usually people who qualify have severe pain that is not responding to therapy, severe heart disease, severe kidney disease, severe lung problems, strokes impacting their thought processes, or are not able to walk or stand. If someone living with Fabry disease feels that they are unable to work, there are guidelines to help them apply for disability. First time claims are frequently declined, but working with a disability lawyer on appeal may reverse that decision.
For more information about disability benefits, please refer to the Social Security Administration website. The Fabry support groups: Fabry Support and Information Group and National Fabry Disease Foundation also may have suggestions and tips for how best to apply for disability due to Fabry related complications.
Can I combine ERT with other Fabry disease treatments like Galafold?
Theoretically, combining chaperone therapy with enzyme replacement therapy (ERT) should stabilize the ERT enzyme and help it enter the lysosome to break down stored material. However, the timing, safety, and effectiveness of treating Fabry disease with both ERT and chaperone therapy has not been established. In addition, access to both could be difficult as insurance will probably not pay for both medications to be used in one individual as it would be considered experimental. Always speak with your Fabry disease specialist before starting or switching to a new treatment.
As a woman, can I have health problems related to Fabry disease?
Women who have a disease causing change or mutation in one of their GLA genes may experience any or all of the symptoms of Fabry disease. Accordingly, the terminology "carrier" to apply to a woman with Fabry is now considered incorrect. Symptoms of Fabry disease vary more in woman than they do in men due to the inheritance pattern of the condition.
Fabry disease is passed through families in an X-linked inheritance pattern, meaning the GLA gene that causes Fabry disease is located on the X-chromosome. Women have two copies of the X-chromosome (XX) and men have one copy of the X-chromosome and one copy of a Y-chromosome (XY). If men inherit an X chromosome containing the nonworking GLA gene, they are unable to produce alpha-gal enzyme. Without alpha-Gal, GL-3 (Gb3) builds up in the body and causes the symptoms and health problems of Fabry disease. On the other hand, if women inherit an X chromosome with a nonworking or GLA gene they have a second X-chromosome with a working GLA gene that can produce some alpha-Gal enzyme. In the past, it was believed that women who were "carriers" would not have Fabry related health problems because they had a normal second copy of the gene. However, in Fabry disease, unlike many other X-linked disorders, women are not just carriers and they may present with Fabry-related health problems. In some cases, women can have health problems as severe as their male relatives. Since women who carry one copy of a nonworking gene do have symptoms of Fabry disease, it is important that they discuss Fabry disease with their doctor and obtain appropriate referrals to monitor their health. To find a Fabry disease specialist or more information on Fabry disease treatment, please refer to the National Fabry Disease Foundation online resource.
Are there other names for Fabry disease?
Fabry disease can also be referred to as:
Are there good support groups for Fabry disease?
There are several excellent support groups for Fabry disease in the United States and around the world. Support groups are often the best way to find other living with Fabry disease and a great source for up-to-date and accurate information on clinical trials, resources, and treatment. A few key support and advocacy groups are listed below:
Are there any resources in the form of books that can help me learn more about Fabry disease?
The teenage years are a time when teens often begin separating their thoughts, goals, and self-image from those of their parents. They begin exploring who they are and who they want to become as they enter adulthood. For people living with a chronic disease, like Fabry disease, the teen years should also include a gradual transfer of medical care responsibility from parents to the teens themselves. It’s a lot easier to learn to do this slowly and with a plan, rather than jumping headfirst into a fast and confusing crash course of medical needs on their 18th birthday. There are programs and exercises designed to help teens figure out how much they already are managing their own healthcare and how to uncover what they still need to learn and master to successfully manage their own healthcare.
The following book can be obtained from Amazon.com and is a great resource for teens at a time of transition when they have to manage their own healthcare. Written by a genetic counselor and a clinical pharmacist, this great resource is in a workbook format and contains lots of tips for a successful transition. Transitions- What Every Teen Living with a LSD Needs to Know
Are home infusions appropriate for children with Fabry disease?
Home infusions can be an excellent option for children who have Fabry disease and are receiving enzyme replacement therapy (ERT). Often, home infusions are less disruptive to a family schedule and can allow children to attend a full day of school.
In order to learn if a specific child can have ERT infusions at home, talk with the medical team that manages the child’s prescriptions. The medical team will usually consider these four key factors:
Are genetic test results for Fabry disease private?
Some people may feel concerned about maintaining the privacy of their genetic information. Medical doctors aren’t allowed to tell anyone that an individual has had a genetic test or its results without consent; however, health insurance companies can request your medical records after permission is given. Individuals with privacy concerns about their genetic information should discuss these with their genetic counselor or Fabry doctor.